Computational Electrosynthesis
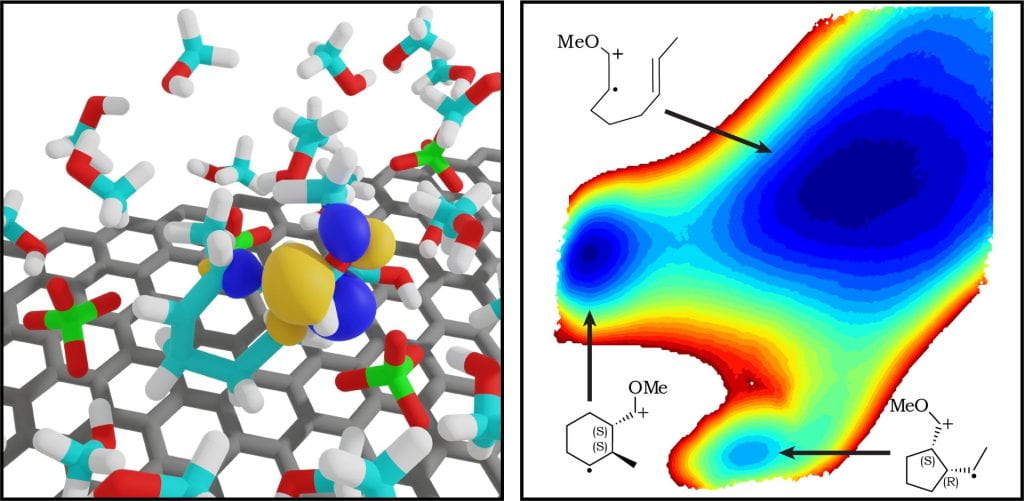
Electrosynthesis can provide new pathways for synthesizing organic molecules, with the working electrode serving as a tunable oxidizing/reducing agent. In many cases, the reactive electrogenerated species are radical ions, which may exhibit unique reactivity characterized by low-barrier bond formation and strong Coulombic interactions with the surrounding electrostatic environment. Our group works to develop and apply computational methods to characterize environmental effects on the reactivity of electrogenerated radical ions, with the goal of rationalizing and predicting trends in yield and selectivity of electrosynthesis applications. Because the initiating electron transfer occurs within the electrical double layer region near the electrode surface, we focus on accurate computational modeling of the atomistic structure of the electrical double layer, under working potentials. We utilize DFT-QM/MM methods to compute free energies of chemical reactions within the electrical double layer, as well as classical molecular dynamics methods to compute additional electrochemical parameters such as redox potentials, reorganization energies, and work terms. Our goal is to provide detailed mechanistic understanding and design rules that lead to development of new electrosynthesis approaches, and better optimization of current electrosynthesis protocols.
Representative Publications:
https://doi.org/10.1021/acs.jpcc.3c03855
Multi-Scale Quantum Chemical Modeling
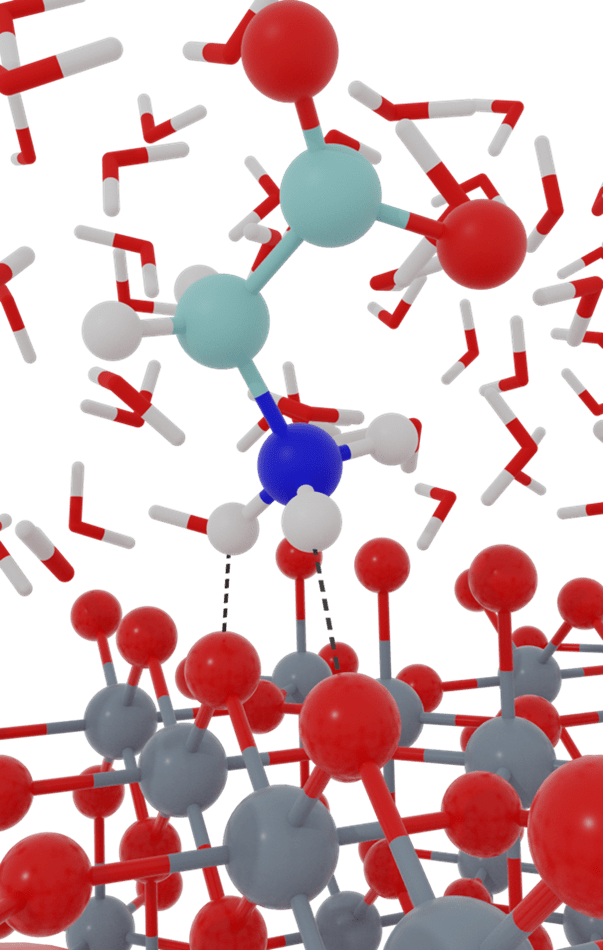
There are many applications in which chemical reactions are substantially modulated by the surrounding environment. Often, the system complexity precludes direct application of quantum chemical methods to the entire system, but rather multi-scale modeling approaches must be employed. Our group works to develop algorithms and software for multi-scale modeling approaches that can be utilized for computing chemical reaction free energies within condensed phase systems, with explicit atomistic representation of the full system. These methods include density functional theory (DFT) based quantum mechanics/molecular mechanics (DFT-QM/MM), quantum embedding approaches, and force field development. We then couple these methods with free energy sampling approaches to investigate mechanisms of chemical reactions in condensed phase environments. An ongoing goal is to advance computational approaches for calculating reaction free energies at solid/liquid interfaces, which has a plethora of applications in heterogeneous catalysis and electrochemistry, but is computationally challenging.
Representative Publications:
https://doi.org/10.1063/5.0087386
Electrochemical Interfaces
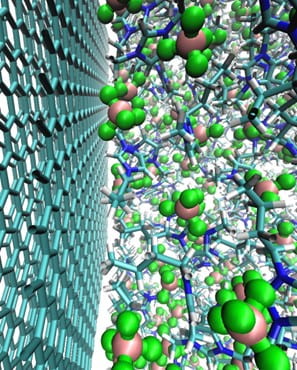
The interface between electrodes and electrolytes hosts a fascinating array of chemical processes that are dictated by voltage-modulated redox chemistry. Numerous chemical and physical processes may be highly coupled and depend sensitively on the microscopic properties of the electrochemical interface, which can change dramatically under applied voltage. Furthermore, depending on the particular device operating conditions, chemical processes may be either thermodynamically or kinetically controlled. We are developing new multi-scale modeling approaches to predict electron transfer, redox chemistry, and chemical reactivity at electrochemical interfaces. Our computational approaches integrate quantum chemical methods and molecular simulation in novel ways, enabling first-principles property prediction through the use of ab initio force fields. We seek to connect with experimental electrochemical, spectroscopic, and scattering measurements to enable comprehensive understanding of these systems.
Representative Publications:
https://doi.org/10.1021/acs.jpcc.2c04910
https://doi.org/10.1021/acs.jpcc.1c05698
Ionic Liquids
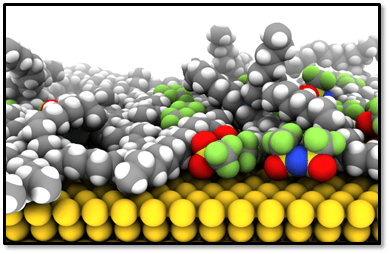
Our group works on various projects related to the physical chemistry and statistical mechanics of room-temperature ionic liquids and organic electrolytes comprised of low-dielectric solvents, for which strong ion-correlation renders mean-field theories entirely inapplicable. We are interested in the phase behavior of ionic liquid mixtures, and also chemical reactions within ionic liquids and other concentrated ionic media. The very large cohesive energy and Coulombic interactions within ionic liquids has a profound effect on the physico-chemical properties within these systems. We are also interested in how ionic liquids and concentrated electrolytes behave in confinement, at interfaces, and under strong applied electric fields for which linear response theories may or may not be valid.
Representative Publications: